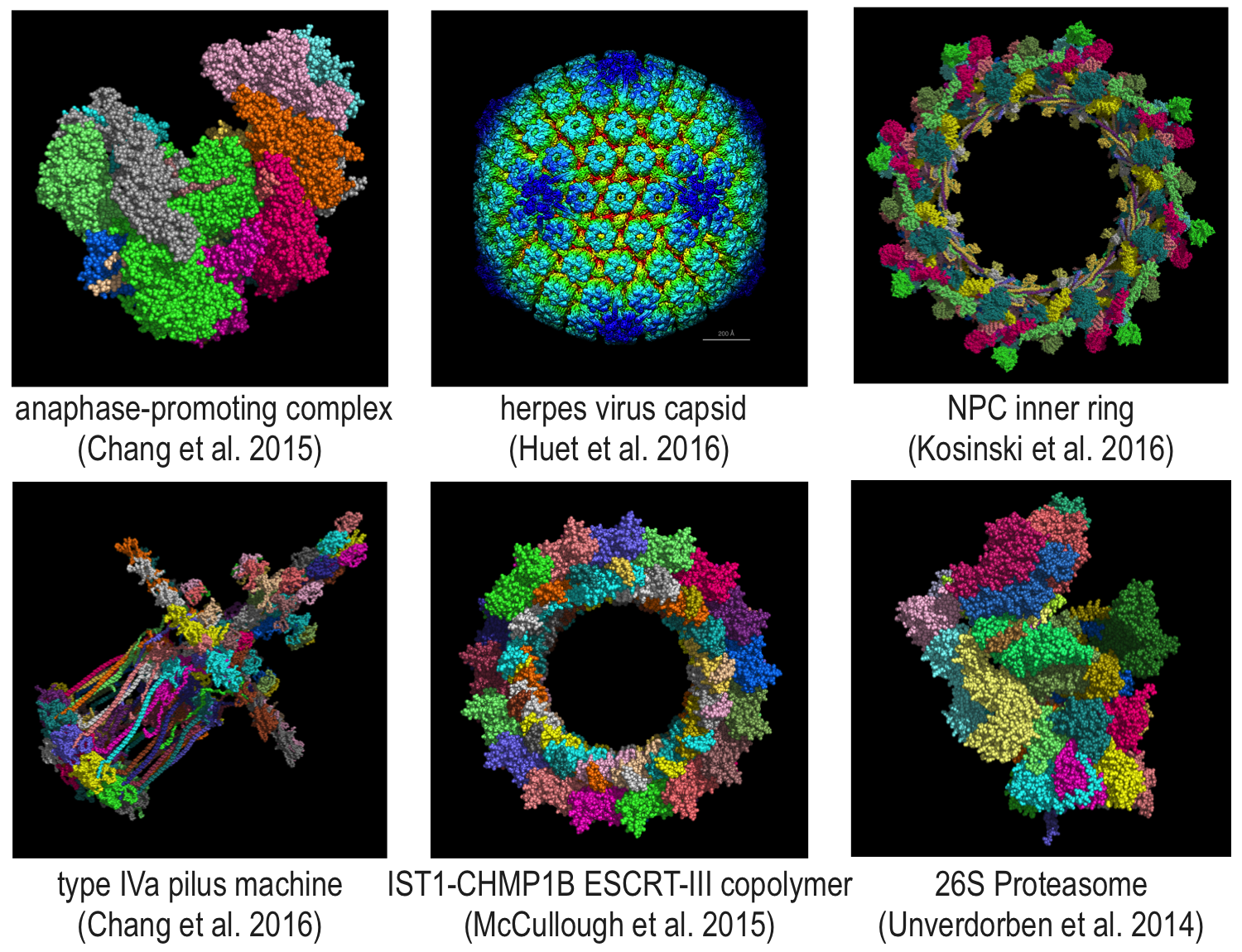
Why structure?
Structural models can aid in:- understanding how cellular machines function
- explaining the phenotype of mutations
- studying the evolution of protein machineries
- designing drug molecules that can control or modify function
- designing new proteins with defined function
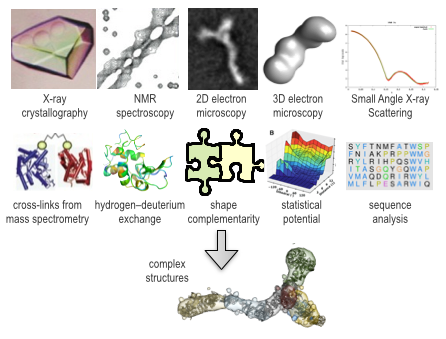
Integrative approach
X-ray crystallography or NMR spectroscopy provide atomic resolution structures but the data collection process is typically long and the success rate is low. Computational methods for modeling assembly structures from individual components frequently suffer from high false-positive rate, rarely resulting in a unique solution.Integrative approach computationally integrates information from experimental methods, bioinformatics, physics, and statistics for rapid and accurate structure determination of protein complexes.
- We focus on development of algorithms that integrate information from any possible source, such as X-ray crystallography, NMR spectroscopy, 2D and 3D Electron Microscopy (EM), Small Angle X-ray Scattering (SAXS), Mass Spectrometry (MS), Hydrogen–deuterium exchange (HDX), mutations, sequence conservation and covariation, and statistical analysis of known structures
- We rely on methods from a wide range of fields, such as computer vision, image processing, computational geometry, machine learning, robotics, and graph algorithms
Dynamics modeling
Dynamics modeling is required to answer the following questions:- which structural states exist for a give protein?
- what is the population of each state?
- what is the transition pathway between different states?
- sampling of the relevant states
- enumeration of possible state combinations (multi-state models)
- scoring of multiple states against the data
- determination of population weights for each of the states
- determination of the relevant number of states

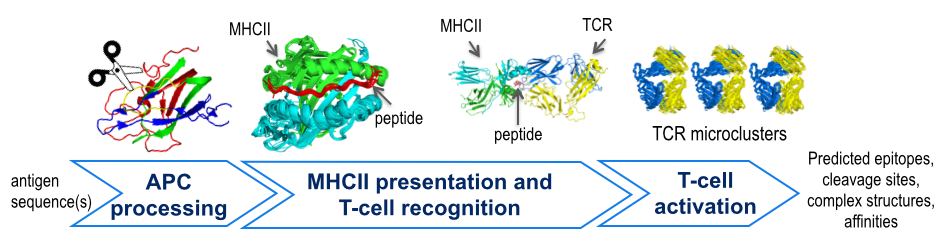
MODELING IMMUNE RESPONSE
Immunogenicity prediction is important for designing vaccines, immunotherapies for cancer and autoimmune diseases, and improved protein therapies. Current in vitro and in vivo methods for predicting immune system epitopes are resource intensive and relatively slow. Computational methods could in principle provide a complementary, rapid and cost-effective approach to assessing protein immunogenicity. We focus on structural modeling of four stages in the immune response pathway:- antigen cleavage - prediction of protease-substrate complex structures and specificities
- MHCII presentation - prediction of peptide-MHCII complex structures and specificities
- TCR recognition - prediction of peptide–MHC–T-cell receptor (TCR) ternary complexes and specificities
- T-cell activation - prediction of structures of signaling microclusters